



Next: About this document ...
Up: main
Previous: Bibliography
Contents
- 1
-
Prat-Resina, X., Garcia-Viloca, M., González-Lafont, A., Lluch, J. M.
On the modulation of the substrate activity for the racemization
catalyzed by mandelate racemase enzyme. a qm/mm study.
Phys. Chem. Chem. Phys. 4:5365-5371, 2002.
- 2
-
Prat-Resina, X., Garcia-Viloca, M., Monard, G., González-Lafont, A., Lluch,
J. M., Anglada, J. M., Bofill, J. M.
The search for stationary points on a quantum mechanical/molecular
mechanical potential-energy surface.
Theor. Chem. Acc. 107:147-153, 2002.
- 3
-
Prat-Resina, X., Bofill, J. M., González-Lafont, A., Lluch, J. M.
Geometry optimization and transition state search in enzymes:
Different options in the micro-iterative method.
Int. J. Quant. Chem. 98(4):367-377, 2004.
- 4
-
Prat-Resina, X., González-Lafont, A., Lluch, J. M.
How important is the refinement of transition state structures in
enzymatic reactions?
J. Mol. Struct. (Theochem) 632:297-307, 2003.
- 5
-
Monard, G., Prat-Resina, X., González-Lafont, A., Lluch, J. M.
Determination of enzymatic reaction pathways using qm/mm methods.
Int. J. Quant. Chem. 93:229-244, 2003.
- 6
-
Nam, K., Prat-Resina, X., Garcia-Viloca, M., Devi-Kesavan, L. S., Gao, J.
Dynamics of an enzymatic substitution reaction in haloalkane
dehalogenase.
J. Am. Chem. Soc. 126:1369-1376, 2004.
- 7
-
Prat-Resina, X., González-Lafont, A., Lluch, J. M.
Free energy calculations on different reaction coordinates of
mandelate racemase.
in preparation.
- 8
-
Pauling, L., Wilson, E. B. Introduction to Quantum Mechanics. With Applications
to Chemistry.
New York: Dover. 1985.
- 9
-
Pilar, F. L.
Elementary Quantum Chemistry: McGraw-Hill Inc.. 1968.
- 10
-
Daudel, R., Leroy, G., Peters, D., Sana, M. Quantum Chemistry.
New York: John Wiley & Sons. 1983.
- 11
-
Jensen, F. Introduction to Computational Chemistry.
West Sussex, England: John Wiley & Sons. 1999.
- 12
-
Fletcher, R. Practical methods of optimization.
2nd Ed. Tiptree, Essex, United Kingdom: John Wiley & Sons. 1987.
- 13
-
Leach, A. R. Molecular Modelling. Principles and Applications.
2nd edition Ed. Essex, England: Pearson Education. 2001.
- 14
-
Schlick, T. Molecular modeling and simulation. An Interdisciplinary Guide.
New York: Springer. 2002.
- 15
-
Allen, M. P., Tildesley, D. J. Computer Simulation of Liquids.
Oxford: Oxford University Press. 1987.
- 16
-
McQuarrie, D. A. Statistical Mechanics.
Sausalito, California: University Science Books. 2000.
- 17
-
Nye, M. J.
From Chemical Philosophy to Theoretical Chemistry: Dynamics of Matter
and Dynamics of Disciplines, 1800-1950: University of California Press. 1994.
- 18
-
Levine, I. N. Química Cuántica.
Spanish version from the original "quantum chemistry" Ed. Madrid: AC.
1977.
- 19
-
Cramer, C. J.
Essentials of Computational Chemistry : Theories and Models: John
Wiley & sons. 2002.
- 20
-
Nakamura, H. Nonadiabatic Transition: Concepts, Basic Theories and
Applications.
Singapore: World Scientific. 2002.
- 21
-
Baer, M.
Introduction to the theory of electronic non-adiabatic coupling terms
in molecular systems.
Phys. Rev. 358:75-142, 2002.
- 22
-
Handy, N. C., Yamaguchi, Y., Schaefer III, H. F.
The diagonal correction to the born-oppenheimer approximation: Its
effect on the singlet-triplet splitting of ch and other molecular
effects.
and other molecular
effects.
J. Chem. Phys. 84(8):4481-4484, 1986.
- 23
-
Wilson, E., Decius, J. C., Cross, P. Molecular Vibrations.
New York: Dover. 1980.
- 24
-
Kosloff, R.
Time-dependent quantum-mechanical methods for molecular dynamics.
J. Phys. Chem. 92:2087-2100, 1988.
- 25
-
Miller, W. H.
The semiclassical initial value representation: A potentially
practical way for adding quantum effects to classical molecular dynamics
simulations.
J. Phys. Chem. A 105:2942-2955, 2001.
- 26
-
Beck, M., JaKckle, A., Worth, G., Meyer, H.-D.
The multiconfiguration time-dependent hartree (mctdh) method: a
highly eficient algorithm for propagating wavepackets.
Phys. Rep. 324:1-105, 2000.
- 27
-
Issue dedicated to time-dependent quantum molecular dynamics.
J. Phys. Chem. A 103(47).
- 28
-
Szabo, A., Ostlund, N. S. Modern Quantum Chemistry.
New York: Dover. 1983.
- 29
-
Goedecker, S.
Linear scaling electronic structure methods.
Rev. Mod. Phys. 71(4):1085-1123, 1999.
- 30
-
Dewar, M. J. S., Thiel, W.
Ground states of molecules, 38. the mndo method. approximations and
parameters.
J. Am. Chem. Soc. 99:4899-4907, 1977.
- 31
-
Dewar, M. J. S., Zoebisch, E. G., Healy, E. F., Stewart, J. J. P.
Am1: A new general purpose quantum mechanical model.
J. Am. Chem. Soc. 107:3902-3909, 1985.
- 32
-
Stewart, J. J. P.
Optimization of parameters for semiempirical methods. i. method.
J. Comp. Chem. 10:209-220, 1989.
- 33
-
Stewart, J. J. P.
Optimization of parameters for semiempirical methods. ii.
applications.
J. Comp. Chem. 10:221-264, 1989.
- 34
-
Clark, T.
Quo vadis semiempirical mo-theory?
J. Mol. Struct. (Theochem) 530:1-10, 2000.
- 35
-
Pople, J. A., Beveridge, D. L. Approximate Molecular Orbital Theory.
New York: McGraw-Hill. 1970.
- 36
-
Bernal-Uruchurtu, M. I., Ruiz-Lopez, M. F.
Basic ideas for the correction of semiempirical methods describing
h-bonded systems.
Chem. Phys. Lett 330:118-124, 2000.
- 37
-
Hohenberg, P., Kohn, W.
Inhomogeneous electron gas.
Phys. Rev. 136:864, 1964.
- 38
-
Ziegler, T.
Approximate density functional theory as a practical tool in
molecular energetics and dynamics.
Chem. Rev. 91:651-667, 1991.
- 39
-
Kohn, W.
Nobel lecture: Electronic structure of matter wave functions and
density functionals.
Rev. Mod. Phys. 71(5):1253-1266, 1999.
- 40
-
Kohn, W., Sham, L.
Self-consistent equations including exchange and correlation effects.
Phys. Rev. A 140:1133, 1965.
- 41
-
Parr, R. G., Yang, W.
Density Functional Theory: Oxford University Press. 1989.
- 42
-
Becke, A. D.
Density-functional exchange-energy approximation with correct
asymptotic behavior.
Phys. Rev. A 38:3098-3100, 1988.
- 43
-
Lee, C., Yang, W., Parr, R. G.
Development of the colle-salvetti correlation-energy formula into a
functional of the electron density.
Phys. Rev. B 37:785-789, 1988.
- 44
-
Becke, A. D.
Density-functional thermochemistry. iii. the role of exact exchange.
J. Chem. Phys. 98(7):5648-5652, 1993.
- 45
-
Koch, W., Holthausen, M. C. Chemist's Guide to Density Functional Theory.
Weinheim: Wiley-VCH. 2000.
- 46
-
Luchow, A., Anderson, J. B.
Monte Carlo methods in electronic structures for large systems.
Annu. Rev. Phys. Chem. 51:501-526, 2000.
- 47
-
Head-Gordon, M.
Quantum chemistry and molecular processes.
J. Phys. Chem. 100(31):13213-13225, 1996.
- 48
-
Steinbach, P. J., Brooks, B. R.
New spherical-cut-off methods for long-range forces in macromolecular
simulation.
J. Comput. Chem 15:667-683, 1994.
- 49
-
Feller, S. E., Pastor, R. W., Rojnuckarin, A., Bogusz, S., Brooks, B. R.
Effect of electrostatic force truncation on interfacial and transport
properties of water.
J. Phys. Chem. 100:17011-17020, 1996.
- 50
-
Frenkel, D., Smit, B. Understanding Molecular Simulation: From Algorithms to
Applications.
San Diego. CA: Academic Press. 1996.
- 51
-
Weiner, S. J., Kollman, P. A., Case, D. A., Singh, U. C., Ghio, C., Alagona,
G. S., Profeta, J., Weiner, P.
-.
J. Am. Chem. Soc. 106:765, 1984.
- 52
-
Pearlman, D., Case, D., Caldwell, J., Ross, W., Cheatham, T., Debolt, S.,
Ferguson, D., Seibel, G., Kollman, P.
Amber, a package of computer-programs for applying molecular
mechanics, normal-mode analysis, molecular- dynamics and free-energy
calculations to simulate the structural and energetic properties of
molecules.
Comp. Phys. Comm. 91(1-3):1-41, 1995.
- 53
-
Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan,
S., Karplus, M.
Charmm: A program for macromolecular energy, minimization and
dynamics calculations.
J. Comput. Chem. 4(2):187-217, 1983.
- 54
-
MacKerell Jr., A. D., Bashford, D., Bellott, M., Dunbrack Jr., R. L.,
Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S.,
Joseph-McCarthy, D., Kuchnir, L., Kuczera, K., Lau, F. T. K., Mattos, C.,
Michnick, S., Ngo, T., Nguyen, D. T., Prodhom, B., Reiher III, W. E., Roux,
B., Schlenkrich, M., Smith, J. C., Stote, R., Straub, J., Watanabe, M.,
Wiórkiewicz-Kuczera, J., Yin, D., Karplus, M.
All-atom empirical potential for molecular modeling and dynamics
studies of proteins.
J. Phys. Chem. B 102:3586-3616, 1998.
- 55
-
Jorgensen, W. L., Maxwell, D. S., Tirado-Rives, J.
Development and testing of the opls all-atom force field on
conformational energetics and properties of organic liquids.
J. Am. Chem. Soc. 118(45):11225-11236, 1996.
- 56
-
Scott, W. R. P., Hünenberger, P. H., Tironi, I. G., Mark, A. E., Billeter,
S. R., Fennen, J., Torda, A. E., Huber, T., Krüger, P., van Gunsteren, W. F.
The gromos biomolecular simulation program package.
J. Phys. Chem. A 103(19):3596-3607, 1999.
- 57
-
Price, D. J., Brooks III, C. L.
Modern protein force fields behave comparably in molecular dynamics
simulations.
J. Comput. Chem. 23(23):1045-1057, 2002.
- 58
-
Cramer, C. J., Truhlar, D. G.
Implicit solvation models: Equilibria, structure, spectra, and
dynamics.
Chem. Rev. 99:2161-2200, 1999.
- 59
-
Roux, B. Computational biochemistry & biophysics.
New York: Marcel Dekker Inc. 2001.
- 60
-
Cui, Q.
Combining implicit solvation models with hybrid quantum
mechanical/molecular mechanical methods: A critical test with glycine.
J. Chem. Phys. 117(10):4720-4728, 2002.
- 61
-
Orozco, M., Luque, F. J.
Theoretical methods for the description of the solvent effect in
biomolecular systems.
Chem. Rev. 100:4187-4225, 2000.
- 62
-
Garcia-Viloca, M., Gao, J., Karplus, M., G.Truhlar, D.
How enzymes work:analysis by modern rate theory and computer
simulations.
Science 303:186-195, 2004.
- 63
-
Matsubara, T., Maseras, F., Koga, N., Morokuma, K.
Application of the new "integrated mo + mm" (imomm) method to the
organometallic reaction pt(pr ) + h (r = h, me, t-bu, and ph).
) + h (r = h, me, t-bu, and ph).
J. Phys. Chem. 100(7):2573-2580, 1996.
- 64
-
Maseras, F., Lledós, A. Computational modeling of homogeneous catalysis.
Dordrecht (Holland): Kluwer. 2002: 1-21.
- 65
-
Sierka, M., Sauer, J.
Finding transition structures in extended systems: A strategy based
on a combined quantum mechanics empirical valence bond approach.
J. Chem. Phys. 112(16):6983-6996, 2000.
- 66
-
Warshel, A., Levitt, M.
Theoretical studies of enzymic reactions: dielectric, electrostatic
and steric stabilization of the carbonium ion in the reaction of lysozime.
J. Mol. Biol. 103:227-249, 1976.
- 67
-
Singh, U. C., Kollman, P. A.
A combined ab initio quantum mechanical and molecular mechanical
method for carrying out simulations on complex molecular systems:
applications to the
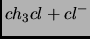 exchange reaction and gas phase protonation
of polyethers.
exchange reaction and gas phase protonation
of polyethers.
J. Comput. Chem. 7(6):718-730, 1986.
- 68
-
Field, M. J., Bash, P. A., Karplus, M.
A combined quantum mechanical and molecular mechanical potential for
molecular dynamics simulations.
J. Comput. Chem. 11(6):700-733, 1990.
- 69
-
Warshel, A., Karplus, M.
Calculation of ground and excited state potential surfaces of
conjugated molecules. i. formulation and parametrization.
J. Am. Chem. Soc. 94(16):5612-5625, 1972.
- 70
-
Gao, J., Thompson, M. A., eds. Combined Quantum Mechanical and Molecular
Mechanical Methods.
ACS Symposium Series 712 Washington D.C.: American Chemical Society.
1998.
- 71
-
Monard, G., Merz, K. M.
Combined quantum mechanical/molecular mechanical methodologies
applied to biomolecular systems.
Acc. Chem. Res. 32:904-911, 1999.
- 72
-
Field, M. J.
Simualting enzyme reactions: challenges and perspectives.
J. Comput. Chem. 23(1):48-58, 2002.
- 73
-
Bakowies, D., Thiel, W.
Hybrid models for combined quantum mechanical and molecular
mechanical approaches.
J. Phys. Chem. 100:10580-10594, 1996.
- 74
-
Besler, B. H., Merz Jr., K. M., Kollman, P. A.
Atomic charges derived from semiempirical methods.
J. Comput. Chem. 11:431-439, 1990.
- 75
-
Luque, F. J., Reuter, N., Cartier, A., Ruiz-Lopez, M. F.
Calibration of the quantum/classical hamiltonian in semiempirical
qm/mm am1 and pm3 methods.
J. Phys. Chem. A 104:10923-10931, 2000.
- 76
-
Cummins, P. L., Gready, J. L.
Coupled semiempirical quantum mechanics and molecular mechanics
(qm/mm) calculations on the aqueous solvation free energies of ionized
molecules.
J. Comput. Chem. 20:1028, 1999.
- 77
-
Freindorf, M., Gao, J.
Optimization of the lennard-jones parameters for a combined ab-initio
quantum-mechanical and molecular mechanical potential using the 3-21g
basis-set.
J. Comput. Chem. 17(4):386-395, 1996.
- 78
-
Martin, M. E., Aguilar, M. A., Chalmet, S., Ruiz-Lopez, M. F.
An iterative procedure to determine lennard-jones parameters for
their use in quantum mechanics/molecular mechanics liquid state simulations.
Chem. Phys. 284:607-614, 2002.
- 79
-
Ranganathan, S., Gready, J. E.
Hybrid quantum and molecular mechanical (qm/mm) studies on the
pyruvate to l-lactate interconversion in l-lactate dehydrogenase.
J. Phys. Chem. B 101(28):5614-5618, 1997.
- 80
-
Reuter, N., Dejaegere, A., Maigret, B., Karplus, M.
Frontier bonds in qm/mm methods: a comparison of different
approaches.
J. Phys. Chem. A 104(8):1720-1735, 1999.
- 81
-
Hall, R. J., Hindle, S. A., Burton, N. A., Hillier, I. H.
Aspects of hybrid qm/mm calculations: the treatment of the qm/mm
interface region and geometry optimization with an application to chorismate
mutase.
J. Comput. Chem. 21(16):1433-1441, 2000.
- 82
-
Antes, I., Thiel, W.
Adjusted connection atoms for combined quantum mechanical and
molecular mechanical methods.
J. Phys. Chem. B 103:9290-9295, 1999.
- 83
-
Cummins, P. L., Gready, J. E.
Combined quantum and molecular mechanics (qm/mm) study of the
ionization state of 8-methylpterin substrate bound to dihydrofolate
reductase.
J. Phys. Chem. B 104(18):4503-4510, 2000.
- 84
-
Zhang, Y., Liu, H., Yang, W.
Free energy calculations on enzyme reactions with an efficient
iterative procedure to determine minimum energy paths on a combined ab initio
qm/mm potential energy surface.
J. Chem. Phys. 112(8):3483-3492, 2000.
- 85
-
Das, D., Eurenius, K. P., Billings, E. M., Sherwood, P., Chatfield, D. C.,
Hodoscek, M., Brooks, B. R.
Optimization of quantum mechanical molecular mechanical partitioning
schemes: Gaussian delocalization of molecular mechanical charges and the
double link atom method.
J. Chem. Phys. 117(23):10534-10547, 2002.
- 86
-
DiLabio, G. A., Hurley, M. M., Christiansen, P. A.
Simple one-electron quantum capping potentials for use in hybrid
qm/mm studies of biological molecules.
J. Chem. Phys. 116(22):9578-9584, 2002.
- 87
-
Swart, M.
Addremove: A new link model for use in qm/mm studies.
Int. J. Quant. Chem. 91:177-183, 2003.
- 88
-
Théry, V., Rinaldi, D., Rivail, J.-L., Maigret, B., Frenczy, B.
Quantum mechanical computations on very large molecular systems : the
local self-consistent field method.
J. Comp. Chem. 15:269-282.
- 89
-
Monard, G., Loos, M., Théry, V., Baka, K., Rivail, J.-L.
Hybrid classical quantum force field for modeling very large
molecules.
Int. J. Quantum Chem. 58:153-159, 1996.
- 90
-
Nicolas Ferré, J.-L. R. Xavier Assfeld.
Specific force field parameters determination for the hybrid ab
initio qm/mm lscf method.
J. Comp. Chem. 23:610-624, 2002.
- 91
-
Gao, J., Amara, P., Alhambra, C., Field, M. J.
A generalized hybrid orbital (gho) method for the treatment of
boundary atoms in combined qm/mm calculations.
J. Phys. Chem. A 12(24):4714-4721, 1998.
- 92
-
Amara, P., Field, M. J., Alhambra, C., Gao, J.
The generalized hybrid orbital (gho) method for combined qm/mm
calculations: Formulation and tests of the analytical derivatives.
Theor. Chem. Acc. 104:336-343, 2000.
- 93
-
Garcia-Viloca, M., Gao, J.
Generalized hybrid orbital for the treatment of boundary atoms in
combined quantum mechanical and molecular mechanical calculations using the
semiempirical parameterized model 3 method.
Theor. Chem. Acc. ASAP.
- 94
-
Pu, J., Gao, J., Truhlar, D. G.
Generalized hybrid orbital (gho) method for combining ab initio
hartree-fock wave functions with molecular mechanics.
J. Phys. Chem. A 108(4):632-650, 2004.
- 95
-
Murphy, R., Philipp, D., Friesner, R.
Frozen orbital qm/mm methods for density functional theory.
Chem. Phys. Lett. 321:113-120, 2000.
- 96
-
Hyperchem.
HyperChem Users Manual.
1998.
- 97
-
Cui, Q., Elstner, M., Kaxiras, E., Frauenheim, T., Karplus, M.
A qm/mm implementation of the self-consistent charge density
functional tight binding (scc-dftb) method.
J. Phys. Chem. B 105:569-585, 2001.
- 98
-
Formaneck, M. S., Li, G., Zhang, X., Cui, Q.
Modeling zinc in biomolecules with the self consistent charge-density
functional tight binding (scc-dftb) method:applications to structural and
energetic analysis.
J. Comp. Chem. 24:565-581, 2003.
- 99
-
Lee, Y. S., Worthington, S. E., Krauss, M., Brooks, B. R.
Reaction mechanism of chorismate mutase studied by the combined
potentials of quantum mechanics and molecular mechanics.
J. Phys. Chem. B 106:12059-12065, 2002.
- 100
-
Lyne, P. D., Hodoscek, M., Karplus, M.
A hybrid qm-mm potential employing hartree-fock or density functional
methods in the quantum region.
J. Phys. Chem. A 103:3462-3471, 1999.
- 101
-
Tuñón, I., Martins-Costa, M., Millot, C., Ruiz-López, M., Rivail, J.
A coupled density functional-molecular mechanics monte carlo
simulation method: The water molecule in liquid water.
J. Comput. Chem. 17(1):19-29, 1996.
- 102
-
Tuñón, I., Martins-Costa, M., Millot, C., Ruiz-López, M.
Molecular dynamics simulations of elementary chemical processes in
liquid water using combined density functional and molecular mechanics
potentials. i. proton transfer in strongly h-bonded complexes.
J. Chem. Phys. 106(9):3633-3642, 1997.
- 103
-
Friesner, R. A., Dunietz, B. D.
Large-scale ab initio quantum chemical calculations on biological
systems.
Acc. Chem. Res. 34:351-358, 2001.
- 104
-
Sherwood, P., de Vries, A. H., Guest, M. F., Schreckenbach, G., Catlow, C.
R. A., French, S. A., Sokol, A. A., Bromley, S. T., Thiel, W., c, A. J. T.,
Billeter, S., Terstegen, F., Thiel, S., Kendrick, J., Rogers, S. C., Casci,
J., Watson, M., King, F., Karlsen, E., Sjøvoll, M., Fahmi, A., Schafer, A.,
Lennartz, C.
Quasi: A general purpose implementation of the qm/mm approach and its
application to problems in catalysis.
J. Mol. Struct. (Theochem) 632:1-28, 2003.
- 105
-
Kongsted, J., Osted, A., Mikkelsen, K. V., Christiansen, O.
Coupled cluster/molecular mechanics method: Implementation and
application to liquid water.
J. Phys. Chem. B 107:2578-2588, 2003.
- 106
-
Car, R., Parrinello, M.
Unified approach for molecular dynamics and density-functional
theory.
Phys. Rev. Lett. 55(22):2471-2474, 1985.
- 107
-
Carloni, P., Rothlisberger, U., Parrinello, M.
The role and perspective of ab initio molecular dynamics in the study
of biological systems.
Acc. Chem. Res. 35:455-464, 2002.
- 108
-
Maseras, F., Morokuma, K.
Imomm: A new integrated ab initio + molecular mechanics geometry
optimization scheme of equilibrium structures and transition states.
J. Comput. Chem. 16(9):1170-1179, 1995.
- 109
-
Dapprich, S., Komáromi, I., Byun, K. S., Morokuma, K., Frisch, M. J.
A new oniom implementation in gaussian98. part i. the calculation of
energies, gradients, vibrational frequencies and electric field derivatives.
J. Mol. Struct. (Theochem) 461:1-21, 1999.
- 110
-
Hurley, M. M., Wright, J. B., Lushington, G. H., White, W. E.
Quantum mechanics and mixed quantum mechanics/molecular mechanics
simulations of model nerve agents with acetylchozlinesterase.
Theor. Chem. Acc. 109:160-168, 2003.
- 111
-
Vreven, T., Morokuma, K.
Investigation of the s s
s excitation in bacteriorhodopsin
with the oniom (mo:mm) hybrid method.
excitation in bacteriorhodopsin
with the oniom (mo:mm) hybrid method.
Theor. Chem. Acc 109(3):125-132, 2003.
- 112
-
Warshel, A.
Computer Modeling of Chemical Reactions in Enzymes and Solutions: New
York. 1992.
- 113
-
Warshel, A.
Molecular dynamics simulations of enzymatic reactions.
Acc. Chem. Res. 35:385-395, 2002.
- 114
-
Warshel, A., Parson, W. W.
Dynamics of biochemical and biophysical reactions : insight from
computer simulations.
Quarterly Review of Biopysics 4:563-679, 2001.
- 115
-
Warshel, A.
Computer simulations of enzyme catalysis: Methods, progress, and
insights.
Ann. Rev. Biophys. Biomol. Struct. 32:425-443, 2003.
- 116
-
Mo, Y., Gao, J.
An ab initio molecular orbital-valence bond (movb) method for
simulating chemical reactions in solution.
J. Phys. Chem. A 104:3012-3020, 2000.
- 117
-
Devi-Kesavan, L. S., Garcia-Viloca, M., Gao, J.
Semiempirical qm/mm potential with simple valence bond (svb) for
enzyme reactions. application to the nucleophilic addition reaction in
haloalkane dehalogenase.
Theor. Chem. Acc. 109(3):133-139, 2003.
- 118
-
Hong, G., Strajbl, M., Wesolowski, T. A., Warshel, A.
Constraining the electron densities in dft method as an effective way
f or ab initio studies of metal-catalyzed reactions.
J. Comp. Chem. 21(16):1554-1561, 2000.
- 119
-
Cummins, P. L., Gready, J. E.
Computational methods for the study of enzymic reaction mechanisms.
ii. an overlapping mechanically embedded method for hybrid
semi-empirical-qm/mm calculations.
J. Mol. Struct. (Theochem) 632:247-257, 2003.
- 120
-
Hayashi, S., Ohmine, I.
Proton transfer in bacteriorhodopsin: Structure, excitation, ir
spectra, and potential energy surface analyses by an ab initio qm/mm method.
J. Phys. Chem. B 104:10678-10691, 2000.
- 121
-
Poteau, R., Ortega, I., Alary, F., Solis, A. R., Barthelat, J.-C., Daudey,
J.-P.
Effective group potentials. 1. method.
J. Phys. Chem. A 105:198-205, 2001.
- 122
-
Kairys, V., Jensen, J. H.
Qm/mm boundaries across covalent bonds: A frozen localized molecular
orbital-based approach for the effective fragment potential method.
J. Phys. Chem A 104:6656-6665, 2000.
- 123
-
Gogonea, V., Westerhoff, L. M., Merz Jr., K. M.
Quantum mechanical/quantum mechanical methods. i. a divide and
conquer strategy for solving the schrodinger equation for large molecular
systems using a composite density functional semiempirical hamiltonian.
J. Chem. Phys. 113(14):5604-5613, 2000.
- 124
-
Cui, Q., Guo, H., Karplus, M.
Combining ab initio and density functional theories with
semiempirical methods.
J. Chem. Phys. 117(12):5617-5631, 2002.
- 125
-
Komeiji, Y., Nakano, T., Fukuzawa, K., Ueno, Y., Inadomi, Y., Nemoto, T.,
Uebayasi, M., G.Fedorov, D., a, K. K.
Fragment molecular orbital method:i application to molecular dynamics
simulation, ab initio fmo-md.
Chem. Phys. Lett. 372:342-347, 2003.
- 126
-
Zhang, D. W., Zhang, J. Z. H.
Molecular fractionation with conjugate caps for full quantum
mechanical calculation of protein molecule interaction energy.
J. Chem. Phys. 119(7):3599-3605, 2003.
- 127
-
Cui, Q., Karplus, M.
Molecular properties from combined qm/mm methods. i. analytical
second derivative and vibrational calculations.
J. Chem. Phys. 112(3):1133-1149, 2000.
- 128
-
Wales, D. J.
A microscopic basis for the global appearance of energy landscapes.
Science 293:2067-2070, 2001.
- 129
-
Onuchic, J. N., Luthey-Schulten, Z., Wolynes, P. G.
Theory of protein folding: The energy landscape perspective.
Annu. Rev. Phys. Chem. 48:545-600, 1997.
- 130
-
Simons, J., Jorgensen, P., Taylor, H., Ozment, J.
J. Phys. Chem. 87:2745, 1983.
- 131
-
Banerjee, A., Adams, N., Simons, J., Shepard, R.
Search for stationary points on surfaces.
J. Phys. Chem. 89:52-57, 1985.
- 132
-
Pulay, P.
Improved scf convergence acceleration.
J. Comput. Chem. 3:556, 1982.
- 133
-
Császár, P., Pulay, P.
Geometry optimization by direct inversion in the iterative subspace.
J. Mol. Struct. 114:31-34, 1984.
- 134
-
Wittbrodt, J. M., Schlegel, H. B.
Estimating stretching force constants for geometry optimization.
J. Mol. Struct. (Theochem) 398:55-61, 1997.
- 135
-
Bofill, J. M.
Updated hessian matrix and the restricted step method for locating
transition structures.
J. Comput. Chem. 15(1):1-11, 1994.
- 136
-
Anglada, J. M., Bofill, J. M.
How good is a broyden-fletcher-goldfarb-shanno-like update hessian
formula to locate transition structures? specific reformulation of
broyden-fletcher-goldfarb-shanno for optimizing saddle points.
J. Comput. Chem. 19(3):349-362, 1998.
- 137
-
Bolhuis, P. G., Chandler, D., Dellago, C., Geissler, P. L.
Transition path sampling: throwing ropes over rough mountain passes,
in the dark.
Annu. Rev. Phys. Chem. 53:291-318, 2002.
- 138
-
Schlegel, B.
Exploring potential energy surfaces for chemical reactions: An
overview of some practical methods.
J. Comput. Chem. 24(12):1515-1527, 2003.
- 139
-
Heidrich, D., ed. The Reaction Path in Chemistry: Current Approaches and
Perspectives.
Dordrecht: Kluwer Academic. 1995.
- 140
-
Eurenius, K. P., Chatfield, D. C., Brooks, B. R., Hodoscek, M.
Enzyme mechanisms with hybrid quantum mechanical and molecular
mechanical potentials. i. theoretical considerations.
Int. J. Quantum Chem. 60(6):1189-1200, 1996.
- 141
-
Fukui, K.
The path of chemical reactions - the irc approach.
Acc. Chem. Res. 14(12):363-368, 1981.
- 142
-
Henkelman, G., Jóhannesson, G., Jónsson, H.
Methods for finding saddle points and minimum energy paths.
In: Progress on Theoretical Chemistry and Physics. Schwartz, S. D.
ed. . Kluwer Academic Publishers 2000 269-300.
- 143
-
Henkelman, G., Jónsson, H.
Improved tangent estimate in the nudged elastic band method for
finding minimum energy paths and saddle points.
J. Chem. Phys. 113(22):9978-9985, 2001.
- 144
-
Chu, J.-W., Trout, B. L., Brooks, B. R.
A super-linear minimization scheme for the nudged elastic band
method.
J. Chem. Phys. 119(24):12708-12717, 2003.
- 145
-
Crehuet, R., Field, M. J.
A temperature-dependent nudged-elastic-band algorithm.
J. Chem. Phys. 118(21):9563-9571, 2003.
- 146
-
Fischer, S., Karplus, M.
Conjugate peak refinement: an algorithm for finding reaction paths
and accurate transition states in systems with many degrees of freedom.
Chem. Phys. Lett. 194(3):252-261, 1992.
- 147
-
Dutzler, R., Schirmer, T., Karplus, M., Fischer, S.
Translocation mechanism of long sugar chains across the maltoporin
membrane channel.
Structure 10(9):1273-1284, 2002.
- 148
-
Woodcock, H. L., Hodoscek, M., Sherwood, P., Lee, Y. S., Schaefer III, H. F.,
R.Brooks, B.
Exploring the quantum mechanical/molecular mechanical replica path
method: a pathway optimization of the chorismate to prephenate claisen
rearrangement catalyzed by chorismate mutase.
Theor. Chem. Acc. 109(3):140-148, 2003.
- 149
-
Pulay, P., Fogarasi, G.
Geometry optimization in redundant internal coordinates.
J. Chem. Phys. 96(4):2856-2860, 1992.
- 150
-
Kudin, K. N., Scuseria, G. E., Schlegel, H. B.
A redundant internal coordinate algorithm for optimization of
periodic systems.
J. Chem. Phys. 114(7):2919-2923, 2001.
- 151
-
Eckert, F., Pulay, P., Werner, H.-J.
Ab initio geometry optimization for large molecules.
J. Comput. Chem. 18(12):1473-1483, 1997.
- 152
-
Nocedal, J.
Updating quasi-newton matrices with limited storage.
Mathematics of Computation 35(151):773-782, 1980.
- 153
-
Liu, D. C., Nocedal, J.
On the limited memory bfgs method for large scale optimization.
Math. Programming 45:503-528, 1989.
- 154
-
Moreales, J. L., Nocedal, J.
Enriched methods for large-scale unconstrained optimization.
Comp. Opt. Appl. 21(2):143-154, 2002.
- 155
-
Anglada, J. M., Besalú, E., Bofill, J. M., Rubio, J.
Another way to implement the powell formula for updating hessian
matrices related to transition structures.
J. Math. Chem. 25:85-92, 1999.
- 156
-
Schlick, T., Overton, M.
J. Comput. Chem. 8:1025, 1987.
- 157
-
Derremaux, P., Zhang, G., Schlick, T., Brooks, B.
A truncated newton minimizer adapted for charmm and biomolecular
applications.
J. Comput. Chem. 15(5):532-552, 1994.
- 158
-
Thomas, A., Field, M. J.
Reaction mechanism of the hgxprtase from plasmodium falciparum:
A hybrid potential quantum mechanical/ molecular mechanical study.
J. Am. Chem. Soc. 12432-12438, 2002.
- 159
-
Moliner, V., Turner, A. J., Williams, I. H.
Transition-state structural refinement with grace and charmm:
realistic modelling of lactate dehydrogenase using a combined
quantum/classical method.
J. Chem. Soc. Chem. Comm. 14:1271-1272, 1997.
- 160
-
Turner, A. J., Moliner, V., Williams, I. H.
Transition-state structural refinement with grace and charmm:
Flexible qm/mm modelling for lactate dehydrogenase.
Phys. Chem. Chem. Phys. 1:1323-1331, 1999.
- 161
-
Billeter, S. R., Turner, A. J., Thiel, W.
Linear scaling geometry optimisation and transition state search in
hybrid delocalised internal coordinates.
Phys. Chem. Chem. Phys. 2:2177-2186, 2000.
- 162
-
Vreven, T., Morokuma, K., Farkas, Ö., Schlegel, H. B., Frisch, M. J.
Geometry optimization with combined methods. i. micro-iterations and
constraints.
J. Comput. Chem. 24(6):760-769, 2003.
- 163
-
Németh, K., Coulaud, O., Monard, G., Ángyán, J. G.
Linear scaling algorithm for the coordinate transformation problem of
molecular geometry optimization.
J. Chem. Phys. 113(6):5598-5603, 2000.
- 164
-
Németh, K., Coulaud, O., Monard, G., Ángyán, J. G.
An efficient method for the coordinate transformation problem of
massively three-dimensional networks.
J. Chem. Phys. 114(22):9747-9753, 2001.
- 165
-
Paizs, B., Baker, J., Suhai, S., Pulay, P.
Geometry optimization of large biomolecules in redundant internal
coordinates.
J. Chem. Phys 113(16):6566-6572, 2000.
- 166
-
Tuckerman, M. E., Martyna, G. J.
Understanding modern molecular dynamics: Techniques and applications.
J. Phys. Chem. B 104:159-178, 2000.
- 167
-
Goldstein, H. Mecánica Clásica.
Barcelona: Reverté. 1996.
Spanish translation of the second english edition.
- 168
-
Truhlar, D. G., Gao, J., Alhambra, C., Garcia-Viloca, M., Corchado, J.,
Sánchez, M. L., Villá, J.
The incorporation of quantum effects in enzyme kinteics modeling.
Acc. Chem. Res. 35(6):341-349, 2002.
- 169
-
Elber, R., Ghosh, A., Cárdenas, A.
Long time dynamics of complex systems.
Acc. Chem. Res. 35:396-403, 2002.
- 170
-
Issue dedicated to molecular dynamics simulations of biomolecules.
Acc. Chem. Res. 35(6).
- 171
-
Andersen, H. C.
Molecular dynamics simulations at constant pressure and/or
temperature.
J. Chem. Phys. 72(4):2384-2393, 1980.
- 172
-
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., Haak,
J. R.
Molecular dynamics with coupling to an external bath.
J. Chem. Phys. 81(8):3684-3690, 1984.
- 173
-
Nosé, S.
A unified formulation of the constant temperature molecular dynamics
methods.
J. Chem. Phys. 81(1):511-519, 1984.
- 174
-
Hoover, W. G.
Canonical dynamics: Equilibrium phase-space distributions.
Phys. Rev. A 31(3):1695-1697, 1985.
- 175
-
Martyna, G. J., and, M. L. K.
Nosé-hoover chains: The canonical ensemble via continuous dynamics.
J. Chem. Phys. 97(4):2635-2643, 1992.
- 176
-
Cheng, A., Merz Jr., K. M.
Application of the nosé-hoover chain algorithm to the study of
protein dynamics.
J. Phys. Chem. 100(5):1927-1937, 1996.
- 177
-
Zhang, Y., Feller, S. E., Brooks, B. R., Pastor, R. W.
Computer simulation of liquid/liquid interfaces. i. theory and
application to octane/water.
J. Chem. Phys. 103(23):10252-10266, 1995.
- 178
-
Ryckaert, J.-P., Ciccotti, G., Berendsen, H. J. C.
Numerical integration of the cartesian equations of motion of a
system with constraints: Molecular dynamics of n-alkanes.
J. Comp. Phys. 23:327-341, 1977.
- 179
-
Kräutler, V., Gunsteren, W. F. V., Hünenberger, P. H.
A fast shake algorithm to solve distance constraint equations for
small molecules in molecular dynamics simulations.
J. Comput. Chem. 22(5):501-508, 2001.
- 180
-
Tobias, D. J., Brooks III, C. L.
Molecular dynamics with internal coordinate constraints.
J. Chem. Phys. 89(9):5115-5127, 1988.
- 181
-
Coluzza, I., Sprik, M., Ciccotti, G., Fom, A.
Constrained reaction coordinate dynamics for systems with
constraints.
Mol. Phys. 101(18):2885-2894, 2003.
- 182
-
Issue dedicated to computational molecular biophysics.
J. Comput. Phys. 1(151).
- 183
-
Brooks III, C. L., Karplus, M.
Deformable stochastic boundaries in molecular dynamics.
J. Chem. Phys. 79(12):6312-6325, 1983.
- 184
-
Brunger, A., Brooks III, C. L., Karplus, M.
Stochastic boundary conditions for molecular dynamics simulations of
st2 water.
Chem. Phys. Lett. 105(5):495-500, 1984.
- 185
-
Brooks III, C. L., Brunger, A., Karplus, M.
Active site dynamics in protein molecules: A stochastic boundary
molecular-dynamics approach.
Biopolymers 24:843-865, 1985.
- 186
-
Brooks III, C. L., Karplus, M.
Solvent effects on protein motion and protein effects on solvent
motion. dynamics of the active site region of lysozyme.
J. Mol. Biol. 208:159-181, 1989.
- 187
-
Alhambra, C., Gao, J.
Hydrogen bonding interactions in the active site of a low molecular
weight protein tyrosine phosphatase.
J. Comput. Chem. 21:1192-1203, 2000.
- 188
-
Li, G., Zhang, X., Cui, Q.
Free energy perturbation calculations with combined qm/mm potentials
complications, simplifications, and applications to redox potential
calculations.
J. Phys. Chem. B 107:8643-8653, 2003.
- 189
-
Garcia-Viloca, M., Alhambra, C., G.Truhlar, D., Gao, J.
Hydride transfer catalyzed by xylose isomerase: Mechanism and quantum
effects.
J. Comp. Chem. 24:177-190, 2003.
- 190
-
Tolman, R. C. The Principles of Statistical Mechanics.
New York: Dover Publications Inc.. 1979.
- 191
-
Chandler, D. Introduction to Modern Statistical Mechanics.
New York: Oxford University Press. 1987.
- 192
-
Kollman, P.
Free energy calculations: Applications to chemical and biochemical
phenomena.
Chem. Rev. 93:2395-2417, 1993.
- 193
-
Masgrau, L., Àngels González-Lafont, Lluch, J. M.
Dependence of the rate constants on the treatment of internal
rotation modes: The reaction oh + chsh  chs + ho as an
example.
chs + ho as an
example.
J. Comput. Chem. 24(6):701-706, 2003.
- 194
-
Garcia-Viloca, M., Alhambra, C., Truhlar, D. G., Gao, J.
Inclusion of quantum-mechanical vibrational energy in reactive
potentials of mean force.
J. Chem. Phys. 114(22):9953-9958, 2001.
- 195
-
Stanton, R. V., Dixon, S. L., Merz, Jr., K. M.
Free energy perturbation calculations within quantum mechanical
methodologies.
In: Computational Approaches to Biochemical reactivity. Naray-Szabo,
G., Warshel, A. eds. . Kluwer Academic Publishers Dordrecht 1997 103-123.
- 196
-
Torrie, G. M., Valleau, J. P.
Nonphysical sampling distributions in monte carlo free energy
estimation: Umbrella sampling.
J. Comput. Phys. 23:187-199, 1977.
- 197
-
González-Lafont, A., Lluch, J. M., Bertrán, J.
Monte carlo simulations of chemical reactions in solution.
In: Solvent Effects and Chemical Reactivity. Tapia, O., Bertrán, J.
eds. Understanding Chemical Reactivity. Kluwer Academic Publishers Dordrecht
1996 125-177.
- 198
-
Kumar, S., Bouzida, D., Swendsen, R. H., Kollman, P. A., Rosenberg, J. M.
The weighted histogram analysis method for free-energy calculations
on biomolecules. i. the method.
J. Comput. Chem. 13(8):1992, 1011-1021.
- 199
-
Boczko, E. M., Brooks III, C. L.
Constant-temperature free energy surfaces for physical and chemical
processes.
J. Phys. Chem. 97:4509-4513, 1993.
- 200
-
Rajamani, R., Naidoo, K. J., Gao, J.
Implementation of an adaptive umbrella sampling method for the
calculation of multidimensional potential of mean force of chemical reactions
in solution.
J. Comput. Chem. 24:1775-1781, 2003.
- 201
-
Roux, B.
The calculation of the potential of mean force using computer
simulations.
Comput. Phys. Commun. 91:275-282, 1995.
- 202
-
Eyring, H.
The activated complex and the absolute rate of chemical reactions.
Chem. Rev. 17(1):65-77, 1935.
- 203
-
Truhlar, D., Garrett, B., Klippenstein, S.
Current status of transition-state theory.
J. Phys. Chem. 100(31):12771-12800, 1996.
- 204
-
Geissler, P. L., Dellago, C., Chandler, D., Hutter, J., Parrinello, M.
Autoionization in liquid water.
Science 291:2121-2124, 2001.
- 205
-
Gao, J., Truhlar, D.
Quantum mechanical methods for enzyme kinetics.
Ann. Rev. Phys. Chem. 53:467-505, 2002.
- 206
-
Himo, F., Siegbahn, P. E. M.
Quantum chemical studies of radical-containing enzymes.
Chem. Rev. 103:2421-2456, 2003.
- 207
-
Noodleman, L., Lovell, T., Han, W.-G., Li, J., Himo, F.
Quantum chemical studies of intermediates and reaction pathways in
selected enzymes and catalytic synthetic systems.
Chem. Rev. 104(2):459-508, 2004.
- 208
-
York, D. M., Lee, T.-S., Yang, W.
Quantum mechanical treatment of biological macromolecules in solution
using linear-scaling electronic structure methods.
Phys. Rev. Lett. 80(22):5011-5014, 1998.
- 209
-
Boero, M., Terakura, K., Tateno, M.
Catalytic role of metal ion in the selection of competing reaction
paths: A first principles molecular dynamics study of the enzymatic reaction
in ribozyme.
J. Am. Chem. Soc. 124:8949-8957, 2002.
- 210
-
Benkovic, S. J., Hammes-Schiffer, S.
A perspective on enzyme catalysis.
Science 301:1196-1202, 2003.
- 211
-
Villà, J., Warshel, A.
Energetics and dynamics of enzymatic reactions.
J. Phys. Chem. B 105:7887-7907, 2001.
- 212
-
Garcia-Viloca, M., González-Lafont, A., Lluch, J. M.
A qm/mm study of the racemization of vinylglycolate catalyzed by
mandelate racemase enzyme.
J. Am. Chem. Soc. 123:709-721, 2001.
- 213
-
Gerlt, J. A., Kozarich, J. W., Kenyon, G. L., Gassman, P. G.
Electrophilic catalysis can explain the unexpected acidity of carbon
acids in enzyme-catalyzed reactions.
J. Am. Chem. Soc. 113(25):9667-9669, 1991.
- 214
-
Mitra, B., Kallarakal, A. T., Kozarich, J. W., Gerlt, J. A., Clifton, J. R.,
Petsko, G. A., Kenyon, G. L.
Mechanism of the reaction catalyzed by mandelate racemase: Importance
of electrophilic catalysis by glutamic acid 317.
Biochemistry 34(9):2777-2787, 1995.
- 215
-
Gerlt, J. A., Gassman, P. G.
An explanation for rapid enzyme-catalyzed proton abstraction from
carbon acids: importance of late transition states in concerted mechanisms.
J. Am. Chem. Soc. 115(24):11552-11568, 1993.
- 216
-
Bearne, S. L., Wolfenden, R.
Mandelate racemase in pieces: Effective concentrations of enzyme
functional groups in the transition state.
Biochemistry 36(7):1646-1656, 1997.
- 217
-
Kenyon, G. L., Gerlt, J. A., Petsko, G. A., Kozarich, J. W.
Mandelate racemase: Structure-function studies of a pseudosymmetric
enzyme.
Acc. Chem. Res. 28:178-186, 1995.
- 218
-
Humphrey, W., Dalke, A., Schulten, K.
Vmd: Visual molecular dynamics.
J. Mol. Graph. 14(1):33-38, 1996.
- 219
-
Li, R., Powers, V. M., Kozarich, J. W., Kenyon, G. L.
Racemization of vinylglycolate catalyzed by mandelate racemase.
J. Org. Chem. 60(11):3347-3351, 1995.
- 220
-
Landro, J. A., Kenyon, G. L., Kozarich, J. W.
Mechanism-based inactivation of mandelate racemase by
propargylglycolate.
Bioorg. Med. Chem. Lett. 2(11):1411-1418, 1992.
- 221
-
Goriup, M., Strauss, U. T., Felfer, U., Kroutil, W., Faber, K.
Substrate spectrum of mandelate racemase part 1: Variation of the
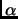 -hydroxy acid moiety.
-hydroxy acid moiety.
J. Mol. Catal. B 15:207-212, 2001.
- 222
-
Whitman, C. P., Hegeman, G. D., Cleland, W. W., Kenyon, G. L.
Symmetry and asymmetry in mandelate racemase catalysis.
Biochemistry 24(15):3936-3942, 1985.
- 223
-
Maurice, M. S., Bearne, S. L.
Reaction intermediate analogues for mandelate racemase: Interaction
between asn 197 and the r-hydroxyl of the substrate promotes catalysis.
Biochemistry 39(44):13324-13335, 2000.
- 224
-
Goriup, M., Strauss, U. T., Felfer, U., Kroutil, W., Faber, K.
Substrate spectrum of mandelate racemase part 2.
(hetero)-aryl-substituted mandelate derivatives and modulation of activity.
J. Mol. Catal. B 15:213-222, 2001.
- 225
-
Maurice, M. S., Bearne, S. L.
The low barrier hydrogen bond in enzymatic catalysis.
J. Biol. Chem. 39(44):13324-13335, 2000.
- 226
-
Schnell, B., Faber, K., Kroutil, W.
Enzymatic racemisation and its application to synthetic
biotransformations.
Adv. Synth. Catal. 345:653-666, 2003.
- 227
-
Kallarakal, A. T., Mitra, B., Kozarich, J. W., Gerlt, J. A., Clifton, J. R.,
Petsko, G. A., Kenyon, G. L.
Mechanism of the reaction catalyzed by mandelate racemase: Structure
and mechanistic properties of the k166r mutant.
Biochemistry 34(9):2788-2797, 1995.
- 228
-
Neidhart, D. J., Howell, P. L., Petsko, G. A., Powers, V. M., Li, R., Kenyon,
G. L., Gerlt, J. A.
Mechanism of the reaction catalyzed by mandelate racemase. 2. crystal
structure of mandelate racemase at 2.5 åresolution: Identification of the
active site and possible catalytic residues.
Biochemistry 30(38):9264-9273, 1991.
- 229
-
Schafer, S. L., Barrett, W. C., Kallarakal, A. T., Mitra, B., Kozarich, J. W.,
Gerlt, J. A.
Mechanism of the reaction catalyzed by mandelate racemase: Structure
and mechanistic properties of the d270n mutant.
Biochemistry 35(18):5662-5669, 1996.
- 230
-
Northrop, D. B.
Follow the protons: A low-barrier hydrogen bond unifies the mechanism
of the aspartic proteases.
Acc. Chem. Res. 34(10):790-797, 2001.
- 231
-
Guthrie, J. P., Kluger, R.
Electrostatic stabilization can explain the unexpected acidity of
carbon acids in enzyme-catalyzed reactions.
J. Am. Chem. Soc. 115(24):11569-11572, 1993.
- 232
-
Alagona, G., Ghio, C., Kollman, P. A.
Ab initio explorative survey of the mechanism catalyzed by mandelate
racemase.
J. Mol. Struct. (Theochem) 390:217-223, 1997.
- 233
-
Landro, J. A., Gerlt, J. A., Kozarich, J. W., Koo, C. W., Shah, V. J., Kenyon,
G. L., Neidhart, D. J., Fujita, S., Petsko, G. A.
The role of lysine 166 in the mechanism of mandelate racemase from
pseudomonas putida: Mechanistic and crystallographic evidence for
stereospecific alkylation by (r)--phenylglycidate.
Biochemsitry 33:635-643, 1994.
- 234
-
Hutter, M. C., Hughes, J. M., Reimers, J. R., Hus, N. S.
Modeling the bacterial photosynthetic reaction center. 2. a combined
quantum mechanical/molecular mechanical study of the structure of the
cofactors in the reaction centers of purple bacteria.
J. Phys. Chem. B 103(23):4906-4915, 1999.
- 235
-
Cheng, A., Stanton, R. S., Vincent, J. J., van der Vaart, A., Damodaran, K. V.,
Dixon, S. L., Hartsough, D. S., Mori, M., Best, S. A., Monard, G.,
Garcia-Viloca, M., Zant, L. C. V., Merz, Jr., K. M. ROAR 2.0.
The Pennsylvania State University. 1999.
- 236
-
Merz Jr., K. M., Banci, L.
Binding of azide to human carbonic anhydrase ii: The role
electrostatic complementarity plays in selecting the preferred resonance
structure of azide.
J. Phys. Chem. 100(43):17414-17420, 1996.
- 237
-
Jorgensen, W. L., Chandrasekhar, J., Madura, J., Impey, R. W., Klein, M. L.
Comparison of simple potential functions for simulating liquid water.
J. Chem. Phys. 79:926-935, 1983.
- 238
-
Gerlt, J. A., Gassman, P. G.
Understanding the rates of certain enzyme-catalyzed reactions: Proton
abstraction from carbon acids, acyl-transfer reactions, and displacement
reactions of phosphodiesters.
Biochemsitry 32(45):11943-11952, 1993.
- 239
-
Frisch, M. J., Trucks, G. W. B., Scuseria, G. E., Robb, M. A., Cheeseman,
J. R., Zakrzewski, V. G., Montgomery Jr., J. A., Stratmann, R. E., Burant,
J. C., Dapprich, S., Millam, J. M., Daniels, A. D., Kudin, K. N., Strain,
M. C., Farkas, O., Tomasi, J., Barone, V., Cossi, M., Cammi, R., Mennucci,
B., Pomelli, C., Adamo, C., Clifford, S., Ochterski, J., Petersson, G. A.,
Ayala, P. Y., Cui, Q., Morokuma, K., Malick, D. K., Rabuck, A. D.,
Raghavachari, K., Foresman, J. B., Cioslowski, J., Ortiz, J. V., Baboul,
A. G., Stefanov, B. B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I.,
Gomperts, R., Martin, R. L., Fox, D. J., Keith, T., Al-Laham, M. A., Peng,
C. Y., Nanayakkara, A., Gonzalez, C., Challacombe, M., Gill, P. M. W.,
Johnson, B., Chen, W., Wong, M. W., Andres, J. L., Gonzalez, C., Head-Gordon,
M., Replogle, E. S., , A., P. J.
Gaussian 98, Revision A.9, Gaussian, Inc., Pittsburgh, PA, USA.
1998.
- 240
-
Schlick, T., Skeel, R., Brunger, A. T., Kalé, L. V., Board, J. A., Hermans,
J., Schulten, K.
Algorithmic challenges in computational molecular biophysics.
J. Comput. Phys. 151:9-48, 1999.
- 241
-
Elber, R., Shalloway, D.
Temperature dependent reaction coordinates.
J. Chem. Phys. 112(13):5539-5545, 2000.
- 242
-
Das, B., Meirovitch, H., Navon, I. M.
Performance of hybrid methods for large-scale unconstrained
optimization as applied to models of proteins.
J. Comput. Chem. 24:1222-1231, 2003.
- 243
-
Anglada, J. M., Besalú, E., Bofill, J. M.
Remarks on large-scale matrix diagonalization using a lagrange-
newton-raphson minimization in a subspace.
Theor. Chem. Acc. 103:163-165, 1999.
- 244
-
Leininger, M. L., Sherrill, C. D., Allen, W. D., Schaefer III, H. F.
Systematic study of selected diagonalization methods for
configuration interaction matrices.
J. Comp. Chem. 22(13):1574-1589, 2001.
- 245
-
Besalú, E., Bofill, J. M.
On the automatic restricted-step rational-function-optimization.
Theor. Chem. Acc. 100:265-274, 1998.
- 246
-
Anderson, E., Bai, Z., Bischof, C., Blackford, S., Demmel, J., Dongarra, J.,
Croz, J. D., Greenbaum, A., Hammarling, S., McKenney, A., Sorensen, D. LAPACK
User's Guide.
3rd Ed. Philadelphia: SIAM. 1999.
- 247
-
Alagona, G., Ghio, C., Kollman, P. A.
Do enzymes stabilize transition states by electrostatic interactions
or pka balance: The case of triose phosphate isomerase (tim)?
J. Am. Chem. Soc. 117(39):9855-9862, 1995.
- 248
-
Andrés, J., Moliner, V., Krechl, J., Silla, E.
J. Chem. Soc. Perkin Trans. 2:1551, 1995.
- 249
-
Gao, J.
Absolute free energy of solvation from monte carlo simulation using
combined quantum and molecular mechanical potentials.
J. Phys. Chem. 96:537-540, 1992.
- 250
-
Truong, T. N., Stefanovich, E. V.
Development of a perturbative approach for monte carlo simulations
using a hybrid ab initio qm/mm method.
Chem. Phys. Lett. 256:348-352, 1996.
- 251
-
Cubero, E., Luque, F. J., Orozco, M., Gao, J.
Perturbation approach to combined qm/mm simulation of solute-solvent
interactions in solution.
J. Phys. Chem. B 107:1664-1671, 2003.
- 252
-
Evans, T. J., Truong, T. N.
Optimizing efficiency of perturbative monte carlo method.
J. Comput. Chem. 19(14):1632-1638, 1998.
- 253
-
Bell, S., Crighton, J. S., Fletcher, R.
A new efficient method for locating saddle points.
Chem. Phys. Lett. 82:122-126, 1981.
- 254
-
Golub, G. H., Loan, C. F. V. Matrix Computations.
3rd Ed. USA: The John Hopkins University Press. 1996.
- 255
-
Li, G., Cui, Q.
Analysis of functional motions in brownian molecular machines with an
efficient block normal mode approach: Myosin-ii and ca -atpase.
-atpase.
Bioph. J. 86(2):743-763, 2004.
- 256
-
Bofill, J. M., de Pinho Ribeiro Moreira, I., Anglada, J. M., Illas, F.
Accurate and efficient determination of higher roots in
diagonalization of larges matrices based in function restricted optimization
algorithms.
J. Comput. Chem. 21(15):1375-1386, 2000.
- 257
-
Bofill, J. M., Anglada, J. M.
Some remarks on the use of the three-term recurrence method in the
configuration interaction eigenvalue problem.
Chem. Phys. 183:19-26, 1994.
- 258
-
Besalú, E., Bofill, J. M.
Calculation of clustered eigenvalues of large matrices using variance
minimization method.
J. Comput. Chem. 19(15):1777-1785, 1998.
- 259
-
Press, W. H., Teukolsky, S. A., Vetterling, W. T., Flannery, B. P. Numerical
Recipes in Fortran 77: The Art of Scientific Computing.
2nd Ed.: Cambridge University Press. copyright 1986-1992.
- 260
-
Gao, J., Garcia-Viloca, M., Poulsen, T. D., Mo, Y.
Solvent effects, reaction coordinates, and reorganization energies on
nucleophilic substitution reactions in aqueous solution.
In: Advances in Physical Organic Chemistry. Vol. 38. Vol. 38. .
Elsevier 2003 161-181.
- 261
-
Gao, J.
A priori computation of a solvent-enhanced sn2 reaction profile in
water: the menshutkin reaction.
J. Am. Chem. Soc. 113(20):7796-7797, 1991.
- 262
-
van Gunsteren, W. F., Mark, A. E.
Validation of molecular dynamics simulation.
J. Chem. Phys. 108(15):6109-6116, 1998.
Xavier Prat Resina
2004-09-09