Models 360
Models 360
(June 2021) This is a standalone version of Models360 and links to ChemEdDL will not work. Xavier Prat-Resina is administering the current version of Models360 and its development, contact him (pratr001@umn.edu) for any question or suggestion about this site.
Molecular Electrostatic Potential (MEP)
Molecular Vibrations
IR spectrum
Click on an IR absorption to see the vibration in the molecular structure.
Symmetry Elements for Point Group C2v
Molecular Orbitals
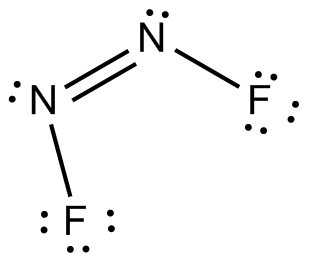
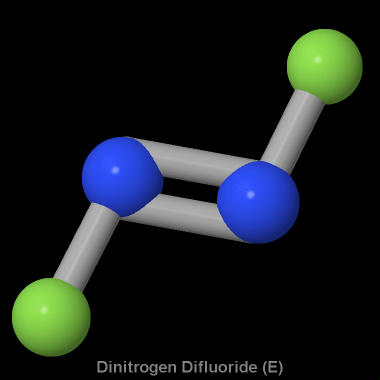
| Smiles: | N(=NF)F |
| Mol. Weight: | 66.01 |
| IUPAC name: | (Z)-difluorodiazene |
| Charge: | 0 |
| InChi: | InChI=1S/F2N2/c1-3-4-2/b4-3- |
| Link to: | Pubchem CID:5364290 |
Additional Information
The molecular structure has been optimized at the B3LYP/6-31g* level of theory. Charges used for electrostatic maps are computed using the NBO method.
The molecular vibrations are the eigenvectors of the Hessian of the potential.
The symmetry group was detected using Serguei Patchkovskii's code (http://www.cobalt.chem.ucalgary.ca/ps/symmetry/)
Most of the Jmol drawing capabilities (Symmetry, Electrostatic Maps, Vibrations and others) are developed by Bob Hanson and the Jmol community of developers.