



Next: Potential of mean force
Up: Introduction: Statistical Mechanics
Previous: Introduction: Statistical Mechanics
Contents
The computation of free energy can be applied to a variety of chemical and biochemical phenomena [192].
Solvation processes, molecular stability, molecular association such as ligand binding ...etc.
We are interested in free energy differences between reactants, products and transition states.
While in gas phase and small molecular systems free energy differences can be computed by means of analytical expressions,
in condensed phase we have to compute the Potential of Mean Force (PMF) along a distinguished coordinate
using sampling techniques.
Free energy calculations for small molecules:
Before describing the PMF calculation in condensed phase systems, it can be useful to indicate the particular case of free energy
calculations in small molecular systems.
In the canonical ensemble (NVT) the Helmholtz free energy 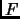 is computed from the canonical partition function
is computed from the canonical partition function 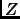
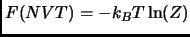 |
(2.114) |
For 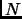 independent and identical particles in the classical limit, is computed from the partition function of a single molecule
independent and identical particles in the classical limit, is computed from the partition function of a single molecule 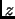 .
.
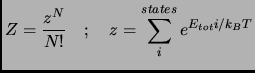 |
(2.115) |
The total energy of every molecular state 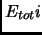 can be approximated as a sum involving translational, rotational,
vibrational and electronic states. This assumption implies that the molecule partition function
can be approximated as a sum involving translational, rotational,
vibrational and electronic states. This assumption implies that the molecule partition function 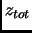 can
be written as a product of terms
can
be written as a product of terms
In small molecular systems the partition functions can be approximated by analytical expressions.
The term  is computed with the free particle model,
is computed with the free particle model, 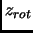 as the rigid rotor and
the
as the rigid rotor and
the 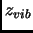 is described as a factorization of normal modes of vibration within the harmonic oscillator.
Some improvements exist for anharmonic and hindered rotor [193] models but in any case
an exploration of the phase space is never performed.
is described as a factorization of normal modes of vibration within the harmonic oscillator.
Some improvements exist for anharmonic and hindered rotor [193] models but in any case
an exploration of the phase space is never performed.
In non-rigid condensed phase systems, the independent particle assumption 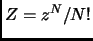 is not valid, and the translational
partition function cannot be modeled as a free particle. Therefore, we will have to compute
the whole partition function through the phase space integral which in the NVT ensemble takes into account all the degrees
of freedom of the configuration space.
is not valid, and the translational
partition function cannot be modeled as a free particle. Therefore, we will have to compute
the whole partition function through the phase space integral which in the NVT ensemble takes into account all the degrees
of freedom of the configuration space.
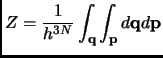 e e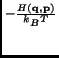 |
(2.117) |
Note that if we consider that our particle system is made up of atoms (nuclei),
the vibrational and translational will be considered altogether.
The usual Hamiltonian expressions permits to separate the atomic momenta from the potential energy.
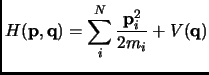 |
(2.118) |
Therefore, the
integral over the phase space 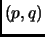 becomes the configurational integral multiplied by a constant
becomes the configurational integral multiplied by a constant
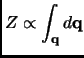 e e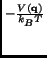 |
(2.119) |
We could consider our condensed phase system (solution or biomolecule) as a supermolecule and apply the same formula
than equations 1.116. However, when computing the vibrational motion of this supermolecule
the local and harmonic approximation is not valid since there are many minima separated by barriers lower than the k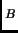 T factor.
In consequence we still need to compute
the internal nuclear motion by MD or MC techniques until convergence of the integral of configuration.
T factor.
In consequence we still need to compute
the internal nuclear motion by MD or MC techniques until convergence of the integral of configuration.
After the assumption of Born-Oppenheimer approximation the nuclei have a classical behavior
and it can be entirely reproduced by classical statistical mechanics.
However, it has been seen that the absence of the zero point energy and the effects of
quantized vibrational motion not contemplated in classical statistical mechanics can be
a source of error, mainly when computing activation free energies in reactions involving hydrogen transfer [194].
Next: Potential of mean force
Up: Introduction: Statistical Mechanics
Previous: Introduction: Statistical Mechanics
Contents
Xavier Prat Resina
2004-09-09