The earliest hybrid method modeling the environment explicitly is the so-called combined Quantum Mechanics/Molecular Mechanics (QM/MM).
In QM/MM [66,67,68]
methodology a small reactive part of a chemical system is
described by quantum mechanics (QM) whereas the remaining large non-reactive
part is described by molecular mechanics (MM).
Although the first paper is attributed to Warshel and Levitt [66] some publications make reference
of a preceding work by Warshel and Karplus [69] where the electronic structure of a polyene
is split in a ![]() system represented by a semiempirical PPP method and the
system represented by a semiempirical PPP method and the ![]() by an empirical force field.
In any case, the method that is used today to address the reactivity of biochemical systems
is still very similar to that used in 1976 by Warshel and Levitt [66].
There is a significant amount of information reviewing the current status of QM/MM methods in the literature
[70,71,72,5].
by an empirical force field.
In any case, the method that is used today to address the reactivity of biochemical systems
is still very similar to that used in 1976 by Warshel and Levitt [66].
There is a significant amount of information reviewing the current status of QM/MM methods in the literature
[70,71,72,5].
![\includegraphics[width=0.4\textwidth]{Figures/qm_split.eps}](img205.png)
|
The main advantage of QM/MM methods is its easy implementation in computational codes while giving rather good chemical results. Its main disadvantage, especially in enzymatic systems, is to go beyond qualitative results and thus to obtain quantitative numbers out of QM/MM computations. This problem is mainly due to three factors: i) the need for good ab initio description for the QM part whereas the usual size of the QM part mostly only allow for semiempirical calculations; ii) the need for accessing free energy numbers through extensive sampling which is too computationally expensive; iii) the difficult calibration of the interaction between the QM part and the MM part, especially in biochemical systems as mentioned there after.
The first two factors are related to actual computational bottlenecks and should be overpassed in the near future.
Splitting the System:
In most reactive systems, the number of atoms involved in a chemical reaction
is fairly limited (i.e., whose electronic properties are changed during
the reaction), the rest of the atoms may have a strong influence
on the reaction but this is usually limited to short and long range
non-bonded interactions that can be represented through both electrostatic
and van der Waals interactions.
The main idea of QM/MM methodology is to split the chemical system into two parts:
the first part is small and described by quantum mechanics, it is called
the quantum part.
The second part is the rest of the system and is
described by molecular mechanics, it is called the classical part.
The full Hamiltonian can therefore be expressed as:
where
![]() is the electronic Hamiltonian describing the quantum atoms
in the Born-Oppenheimer approximation.
It has the same expression that in equations 1.5 and 1.7
in page
is the electronic Hamiltonian describing the quantum atoms
in the Born-Oppenheimer approximation.
It has the same expression that in equations 1.5 and 1.7
in page ![]()
In equation 1.62, the Hamiltonian
![]() describes the classical part.
As we defined in the preceding section, a set of atoms interacting in this part can be seen as a set
of point charges
describes the classical part.
As we defined in the preceding section, a set of atoms interacting in this part can be seen as a set
of point charges ![]() in space interacting through a defined force field.
The usual energy expression for every term of the force field has already been commented in section 1.2.2.1 (page
in space interacting through a defined force field.
The usual energy expression for every term of the force field has already been commented in section 1.2.2.1 (page
![]() ).
).
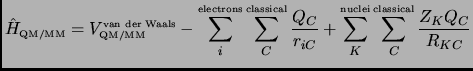
The last term
![]() in equation 1.62
stands for the explicit interaction between the quantum
and the classical part. It can be represented as the sum of two terms: a van der Waals
term describing the non-electrostatic interactions between quantum and classical atoms
and an electrostatic term describing the interaction between a classical point
charge
in equation 1.62
stands for the explicit interaction between the quantum
and the classical part. It can be represented as the sum of two terms: a van der Waals
term describing the non-electrostatic interactions between quantum and classical atoms
and an electrostatic term describing the interaction between a classical point
charge ![]() and the electrons and nuclei of the quantum part:
and the electrons and nuclei of the quantum part:
Note that the second term in equation 1.63
that describes the electrons-classical charge interaction
depends on the coordinates of electrons ![]() . Therefore it must be incorporated into the core
Hamiltonian of the quantum subsystem and obtained through a SCF process.
Indeed this term is the responsible of the polarization on the QM part
due to the presence of the MM charges.
The other two terms in equation 1.63 depend on particles with fixed positions and can
be computed analytically as the rest of MM terms.
. Therefore it must be incorporated into the core
Hamiltonian of the quantum subsystem and obtained through a SCF process.
Indeed this term is the responsible of the polarization on the QM part
due to the presence of the MM charges.
The other two terms in equation 1.63 depend on particles with fixed positions and can
be computed analytically as the rest of MM terms.
QM/MM interactions:
The Hamiltonian
![]() should reproduce quantitatively the interaction between the QM and MM
parts as if the system was computed fully quantum mechanically [73].
The quantitative reproduction of the QM/MM interactions depends on three points:
should reproduce quantitatively the interaction between the QM and MM
parts as if the system was computed fully quantum mechanically [73].
The quantitative reproduction of the QM/MM interactions depends on three points:
1) the choice of the set of non-polarizable point charges ![]() or more
generally of the MM force field;
or more
generally of the MM force field;
2) the choice of the van der Waals parameters
to describe
![]() ;
;
3) the way the classical charges polarize the quantum subsystem.
It is usually a good approximation to take the charge definition from an empirical force field and to incorporate those charges into the core Hamiltonian of the quantum subsystem, being this one the procedure used in this thesis. This gives quite reliable results because the charges in molecular mechanics are defined in order to reproduce electrostatic potential properly [74]. However, it is well-known that between different force fields the charge definition on atoms can change dramatically, even between different generations of the same force field. These differences between different set of charges should have a non-negligible impact on the quality of a QM/MM study.
The choice of van der Waals parameters is another crucial point in the production
of a good QM/MM interaction.
In theory, a new set of parameters should be defined for each atom in the
system (QM + MM) to exclusively compute
![]() [75,76].
It is well known that the parameters from a forcefield are optimized in a consistent fashion.
In consequence the new van der Waals parameters should be compatible with the forcefield used.
In practice, one uses the van der Waals parameters from the current force field
definition for each atom in the MM part and, when possible, define a new set
of van der Waals parameters for the QM atoms [77,78].
[75,76].
It is well known that the parameters from a forcefield are optimized in a consistent fashion.
In consequence the new van der Waals parameters should be compatible with the forcefield used.
In practice, one uses the van der Waals parameters from the current force field
definition for each atom in the MM part and, when possible, define a new set
of van der Waals parameters for the QM atoms [77,78].
A third point needing to be clarified in the description of QM/MM interactions is
the way the classical set of charges ![]() polarize the electronic wavefunction
from the QM subsystem. This is usually done by adding to the core Hamiltonian
of the QM part a perturbation describing the interaction between the QM electrons and the
classical charges. An element of the core Hamiltonian matrix is expressed as:
polarize the electronic wavefunction
from the QM subsystem. This is usually done by adding to the core Hamiltonian
of the QM part a perturbation describing the interaction between the QM electrons and the
classical charges. An element of the core Hamiltonian matrix is expressed as:
 |
(2.64) |
Cutting covalent bonds:
In the study of chemical reactions in solution with QM/MM methods the solute usually belongs
to the quantum part and the solvent to the MM part, so that the two zones are perfectly differentiated.
However, in enzymatic reactions the reactive part treated with QM usually includes the substrate
and some residues that participate actively in the chemical reaction.
In consequence, the frontier between the two zones, mainly in lateral chains of the active aminoacidic
residues resides on a covalent bond.
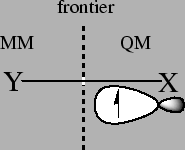
The way this frontier between the two zones is treated has been the objective of many publications. Actually, many laboratories that developed their tools in QM/MM schemes have their own method to treat the frontier
The different solutions can be divided in two main categories. Those that add an atom or pseudoatom to fill the valencies on the quantum frontier, and those which deal specifically with the frontier bond orbital by trying to compute directly its main characteristics from known parameters. In table 1.1 some of the different approaches published so far with its original references is given.
|
The GHO formalism is an extension of the LSCF method which at the same time is derived from the original work from Warshel and Levitt [66]. In GHO the classical frontier atom is described by a set of orbitals divided into two sets of auxiliary and active orbitals. The latter set is included in the SCF calculation, while the former generates an effective core potential for the frontier atom.
As we can see, it seems that there are many variations in QM/MM methods. These methods are still under development and there is not yet a unique and standardized procedure to be used as a black box. Actually, one must face some numerical and methodological problems for every system under study.
More accurate potentials:
Most of publications using QM/MM methods have chosen semiempirical Hamiltonians in the QM part. This is mainly because
an exploration of the configuration space requires thousands of energy and gradient evaluations that so far only
semiempirical methods are fast enough to be permitted.
An interesting initiative is the coupling of a
semiempirical DFT method (SCC-DFTB) with CHARMM package [97],
which is a good compromise between accuracy and computational cost[98].
However, many groups have interfaced ab initio packages to MM programs to perform QM/MM at a higher level of accuracy.
Programs such as Gaussian98, GAMESS-US, GAMESS-UK, CADPAC, HONDO, DALTON, JAGUAR, deMon, Turbomol
are used to carry out
Hartree-Fock/MM[99,100],
DFT/MM[101,102,90,103,104],
or even Coupled Cluster/MM [105].
Finally we must not forget the Carr-Parrinello approximations [106] that has been interfaced with GROMOS force field to perform ab initio simulations of biological systems[107]. Even though, as we have seen, QM/MM methods are far from being accurate. Therefore, in addition to an insufficient sampling, the frontier, the arbitrary polarization charges are sources of error in such a way that the increase of accuracy in the QM part may be in some cases wasted.
![\includegraphics[width=0.3\textwidth]{Figures/gho.eps}](img220.png)