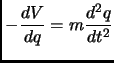Classical molecular dynamics (MD) can be used to propagate in time the nuclear coordinates of molecular system using the classical equations of motion2.10
Equation 1.100 must be solved numerically propagating a trajectory at small time-steps.
While a typical time-step is about one femtosecond (10![]() )
most of chemical interesting events take place at time scales several orders of magnitude higher (micro or millisecond).
Therefore the MD equations should be propagated until 10
)
most of chemical interesting events take place at time scales several orders of magnitude higher (micro or millisecond).
Therefore the MD equations should be propagated until 10![]() -10
-10![]() steps to observe a reactive event (rare event).
Despite of the recent acceleration techniques [169] this task is yet too expensive to be performed in the nowadays computers.
This gap of time scales makes that so far Molecular Dynamics are rarely used
to obtain a real picture of the thermically activated chemical processes.
Therefore, in this thesis MD will be employed as a technique that may be used
to compute equilibrium as well as kinetic properties of a many-body system.
Most of these properties can also be computed with Monte Carlo (MC) techniques.
Although MC covers a vast area
of techniques they will not be commented here, and it may be found in the literature [13,15,50].
steps to observe a reactive event (rare event).
Despite of the recent acceleration techniques [169] this task is yet too expensive to be performed in the nowadays computers.
This gap of time scales makes that so far Molecular Dynamics are rarely used
to obtain a real picture of the thermically activated chemical processes.
Therefore, in this thesis MD will be employed as a technique that may be used
to compute equilibrium as well as kinetic properties of a many-body system.
Most of these properties can also be computed with Monte Carlo (MC) techniques.
Although MC covers a vast area
of techniques they will not be commented here, and it may be found in the literature [13,15,50].
In this section, the main topics of molecular dynamics techniques are presented. However it must be taken in consideration
that MD is a huge field of research and we will only mention those methods that we will apply to model our enzymatic system.
In this sense, a volume of the Account of Chemical Research was dedicated to review the state of the art in
Molecular Dynamics simulations of biomolecules [170].